

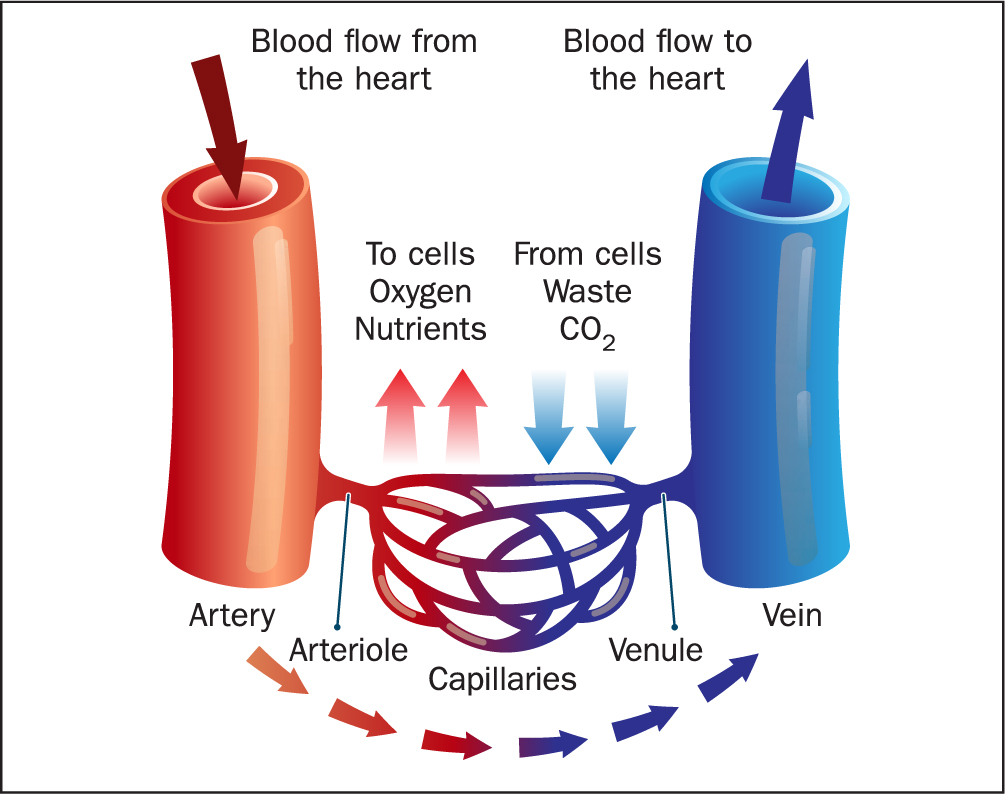
Haematology is a medical specialty dedicated to the study, diagnosis, treatment and prevention of disorders of all blood components. The haematological system consists of blood, blood vessels, bone marrow, lymph nodes, spleen and all proteins involved in haemostasis and thrombosis. Blood circulates the human body through blood vessels (Figure 1), which contain red blood cells, white blood cells and platelets suspended in plasma. Plasma is the liquid component of blood and contains a wide variety of other components, supplying the body with electrolytes, hormones, vitamins, antibodies, oxygen and nourishment, while removing carbon dioxide, nitrogenous waste products and maintaining body temperature (Kumar and Clarke, 2017). Blood tests can be done to measure levels of these and other components in the bloods, and used to aid diagnosis of many diseases when results are outside the normal range.
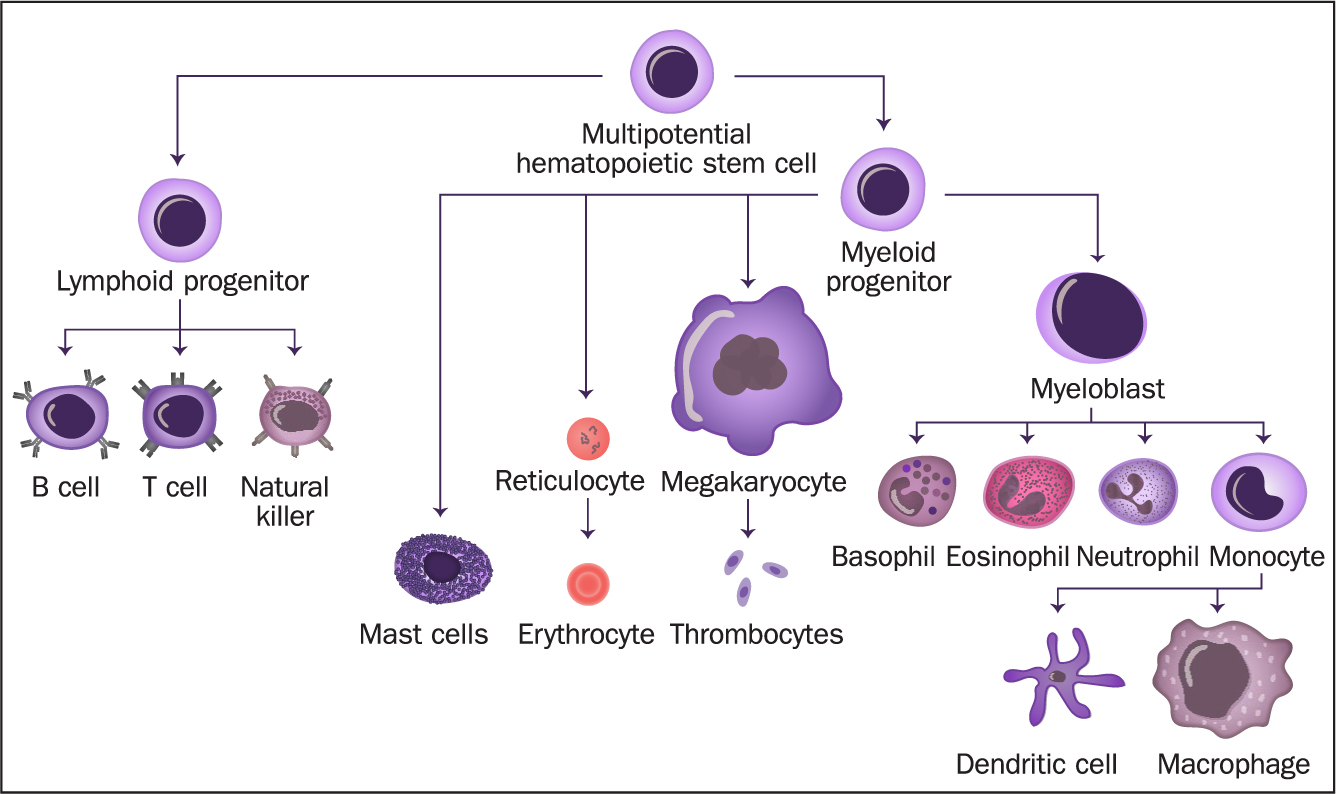
The formation of blood cells (haemopoiesis) takes place in the bone marrow, the spongy centre of most bones. Bone marrow produces multipotent haemopoietic stem cells, meaning they cannot develop into all types of body cell – solely various types of blood cell. These cells specialise further into mature blood cells specifically adapted to their individual roles, via cellular differentiation (Figure 2). This takes place in the liver, the spleen, lymph nodes and thymus, which also help regulate the production and destruction of blood cells (Hoffbrand and Steensma, 2019).
The control of stem-cell activity is important because rapid uncontrolled cellular division allows tumours to grow, whereas reduced cellular division means that tissues are not replaced efficiently, leading to ageing; the last is characterised by a gradual functional decline and is a leading risk factor for cardiovascular disease, diabetes and neurodegenerative disorders, to name a few (McHugh and Gil, 2018). Abnormalities with blood vessels, blood cells and proteins involved in bleeding (haemostasis) and clotting (thrombosis), or any of the organs involved in their development, can also result in disease. Many blood disorders are genetic, although they can also result from other diseases, medicine side-effects or a lack of specific nutrients in the diet, for example vitamin K (Hall, 2016; Carton, 2017).
Blood cell function
White blood cells (leukocytes) are key in fighting infection and ingesting dead cells, tissue debris, mutated cancerous cells and foreign bodies that enter the blood stream, such as allergens via phagocytosis. There are several types of leukocytes: neutrophils, eosinophils, lymphocytes, monocytes and basophils, each type further adapted to its own unique role. Platelets (thrombocytes) are much smaller and are involved in blood clotting to prevent blood loss and pathogens from entering the blood stream through various coagulation pathways.
Red blood cells (erythrocytes) are essential in transporting oxygen from the lungs to respiring cells in the body and taking the waste product, carbon dioxide, back to the lungs to be expired (Hoffbrand and Steensma, 2019). Erythrocytes are adapted for this role by having fewer organelles, to enable them to fit through small capillaries as well as creating more room for haemoglobin (Hb), the protein that binds with oxygen to carry it. Due to this lack of nucleus and organelles erythrocytes are unable to replicate and have a life span of roughly 120 days. Therefore, we produce about 3 billion erythrocytes/kg/day to replace them as required.
Other blood cells also have a high turnover. Platelets, for example, live for about 7 days, while neutrophils survive 6 hours only, requiring the body to produce 1.6 billion neutrophils/kg/hour, proliferating further when fighting infection (Kumar and Clarke, 2017).
Blood group
An individual's blood group is inherited from their parents, and can be identified by antibodies and antigens in the blood (Hall, 2016). Antibodies are proteins in plasma that recognise foreign substances and alert our immune systems to destroy them. Antibodies identify foreign substances by the unique protein molecules (antigens) found on their surface, and there are also antigens on the erythrocytes. There are four blood groups using the ABO system: A, B, AB and O (Table 1). However, erythrocytes can have an additional RhD antigen. Each blood group can therefore be described as either RhD positive or RhD negative, depending on whether the antigen is present or not. This gives a total of eight blood groups.
Table 1. Basic ABO blood group classification
| Blood group | Antigens present on the erythrocytes | Antibodies present in the plasma |
|---|---|---|
| A | A antigens | Anti-B antibodies |
| B | B antigens | Anti-A antibodies |
| O | No antigens | Both anti-A and anti-B antibodies |
| AB | Both A and B antigens | No antibodies |
Due to the presence of antibodies, receiving a blood transfusion from the wrong ABO group can be life-threatening. For example, if an individual with group A blood is given a transfusion of group B blood, their anti-B antibodies will attack the group B cells (Misevic, 2018). Generally, O RhD negative (O-) blood can be safely given to individuals from any other blood group, because these erythrocytes do not have any A, B or RhD antigens on their outer cell membrane. Therefore O-blood is frequently given in medical emergencies before a patient's blood group has been identified (Hall, 2016). In the UK, O is the most common ABO blood group: it is prevalent in 48% of the population, with about 85% of the UK population being RhD positive (NHS website, 2020).
Pregnant women should be tested to identify their blood group. This is because, if the mother is RhD negative but has been exposed to RhD-positive blood (potentially in a previous pregnancy), she develops anti-RhD antibodies; therefore, if the child has inherited RhD-positive blood from the father, the mother's body will immediately produce the anti-RhD antibodies, which will cross the placenta and attack the foetal erythrocytes causing rhesus disease (Visser et al, 2021).
Blood tests
There is a variety of blood tests specific to haematology. For example, full blood count, which measures the size, number and maturity of the various blood cells in the sample (Table 2). Other components of blood can also be measured, including coagulation factors such as Von Willebrand factor (VWF) or factor VIII concentrate (VIII:C) (Palta et al, 2014). Abnormal results in either of these factors will indicate where in the coagulation pathway the problem lies. Bleeding time, prothrombin time (PT) and activated partial thromboplastin time (aPTT) are all clinical laboratory-based tests performed to measure the time it takes for blood to clot. Bleeding time involves creating a standardised incision and timing the cessation of bleeding (Russeau et al, 2021). Conversely, PT involves adding reagents in a laboratory to a blood sample and measuring the time for plasma to clot. The international normalised ratio (INR) was introduced to standardise PT results (Bonar and Favaloro, 2017).
Table 2. Haematology normal adult reference ranges
| Test | Adult range | Units |
|---|---|---|
| Haemoglobin (Hb) | 115–155 in womens135–177 in men | g/L |
| White blood cell count (WBC) | 4.0–11.0 | 109/L |
| Platelets | 150–400 | 109/L |
| Red blood cell count (RBC) | 3.8–5.8 in women4.5–6.5 in men | 1012/L |
| Mean cell volume (MCV) | 80–96 | fL |
| Haematocrit (Hct) | 0.37–0.47 in women0.4–0.54 in men | L/L |
| Mean cell haemoglobin (MCH) | 27–32 | pg |
| Mean cell Hb concentration (MCHC) | 320–360 | g/L |
| Neutrophils | 2.0–7.5 | 109/L |
| Lymphocytes | 1.5–4.0 | 109/L |
| Monocytes | 0.2–0.8 | 109/L |
| Eosinophils | 0.0–0.4 | 109/L |
| Basophils | 0.01–-0.1 | 109/L |
NB Normal values may vary please check with your local laboratory (Kumar and Clark, 2017)
Blood disorders
Although not exhaustive, Table 3 provides some examples of key blood components and associated disorders. All anaemias relate to red blood cells and all leukaemias are cancers that generally relate to white blood cells. Table 4 provides an elaboration of the cause, potential signs and symptoms, as well as diagnosis and management, for a disorder associated with each component.
Table 3. Blood components and associated disorders
| Blood component | Associated disorders |
|---|---|
| Red blood cells |
|
| White blood cells |
|
| Platelets |
|
| Plasma |
|
| Blood group |
|
Table 4. An elaboration of a disorder for each blood component.
| Blood component | Associated disorder | Cause | Potential signs/symptoms | Diagnosis and management |
|---|---|---|---|---|
| Red blood cells | Iron deficiency anaemia | A reduction in haemoglobin (Hb) concentration in the blood due to inadequate iron supply, often due to dietary intake, chronic blood loss from the gut, menstrual periods with abnormally heavy or prolonged bleeding (menorrhagia), or gastrointestinal diseases resulting in the malabsorption of iron |
|
|
| White blood cells | Chronic myelogenous leukaemia | A cancer caused by a genetic mutation in the stem cells produced by the bone marrow, in which translocation between chromosomes 22 and 9 produces a shortened chromosome 22, known as the ‘Philadelphia chromosome’. This genetic mutation results in the stem cells producing too many underdeveloped white blood cells and a reduction in the number of other blood cells, such as red blood cells |
|
|
| Platelets | Immune thrombocytopenic purpura | The body creates platelet auto-antibodies, which contribute to accelerated platelet destruction in the spleen and inhibit platelet production, resulting in a reduced platelet number |
|
|
| Plasma | Deep venous thrombosis | Due to a combination of sluggish blood flow (commonly due to reduced mobility, post-surgery or a long flight) and increased blood coagulability (such as when taking oral contraceptives or smoking), resulting in a blood clot obstructing a vein, most commonly in the legs |
|
|
| Blood group | Transfusion-related acute lung injury (TRALI) | Leucocyte antibodies in the plasma of donors (usually women who have given birth to multiple children) may cause TRALI. These antibodies bind to leucocytes, marking them as foreign bodies which causes an immune response |
|
|
Sickle-cell disease
Sickle-cell disease is an inherited recessive red cell disorder (haemoglobinopathy) that leads to the production of sickle haemoglobin (HbS), instead of just normal adult haemoglobin (HbA), due to a mutation in the β-globin gene (Lobitz et al, 2018). This mutant gene has survived because heterozygote (HbAS) carriers are protected against the effects of severe Plasmodium falciparum malaria (Kariuki and Williams, 2020). However, those with two abnormal genes (HbSS) suffer from sickle-cell disease. Sickle-cell disease is therefore seen most frequently in people of African descent, but is also found in individuals from other areas of current or previous malaria prevalence, such as those with Indian and Arab ancestry.
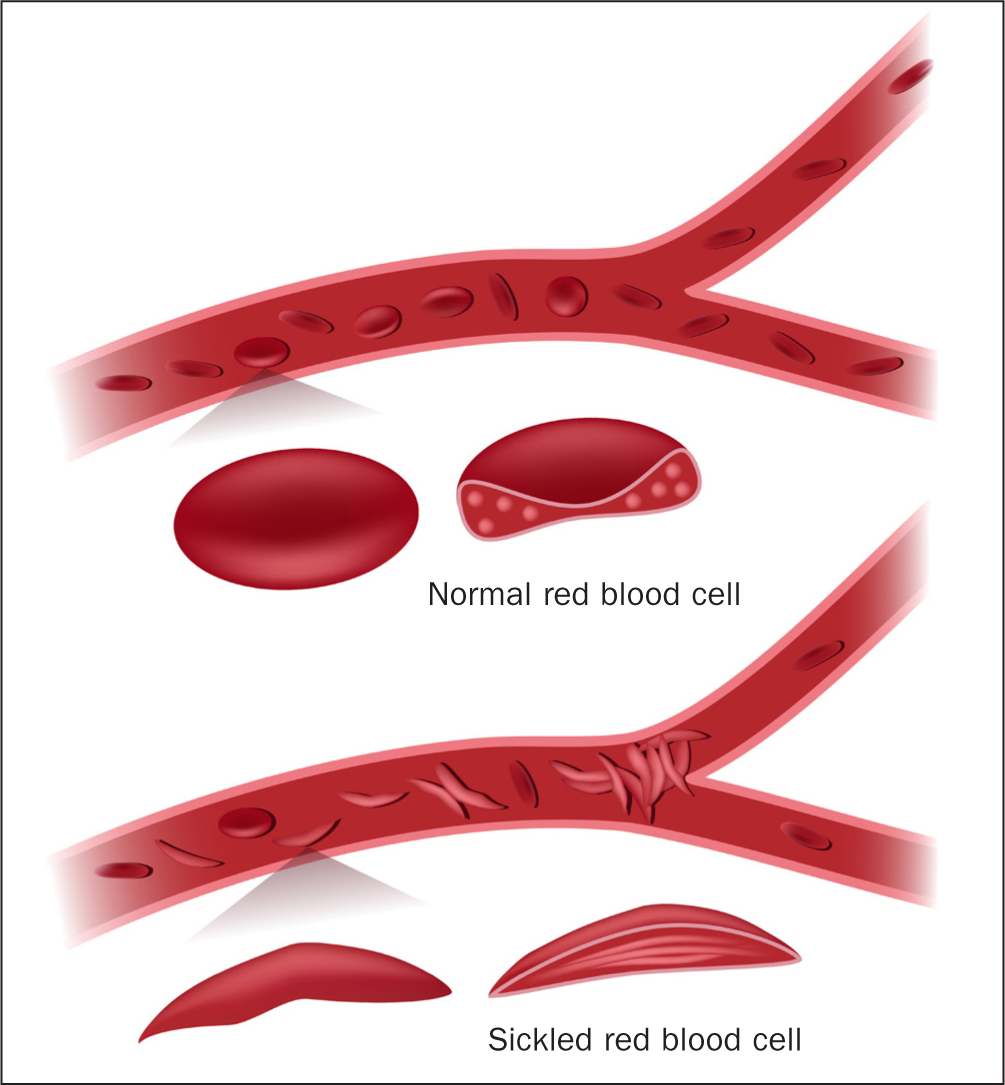
Sickle-cell anaemia is the predominant form of sickle-cell disease, although the proportion varies according to country of origin (Weatherall, 2011). HbS releases its oxygen to the body's tissues more easily than HbA, meaning that individuals do not always realise they are anaemic (Inusa et al, 2019). Deoxygenated HbS molecules in red blood cells are insoluble and polymerise, causing cells to become rigid and adopt the characteristic sickled shape as shown in Figure 3. Sickling can be precipitated by infection, dehydration, acidosis, or sudden cold weather or rain. This process is initially reversible, but with repeated sickling, the cells eventually lose their membrane flexibility and become irreversibly sickled. Sickling can shorten red-cell survival from about 120 days to 10–20 days, as they are more prone to rupturing and releasing their contents into the surrounding plasma, a process called haemolysis. Sickled cells also impair the passage of cells through the smaller blood vessels, leading to obstruction of blood flow (vaso-occlusion), resulting in ischaemia, oedema, pain, necrosis and organ damage (Inusa et al, 2019).
Sickle-cell disease is characterised by both acute and chronic complications presenting when foetal haemoglobin (HbF) drops towards adult level by 6 months of age. Complications range from acute generalised pain (vaso-occlusive crisis), requiring hospitalisation, to early onset stroke, leg ulcers, pulmonary hypertension, haemolytic anaemia (when red blood cells are destroyed faster than they can be made), and the risk of premature death from multi-organ failure (Kato et al, 2018).
In high-income countries more than 94% of children with sickle-cell disease survive to adulthood following early diagnosis, disease education, regular check-ups and preventive therapies such as penicillin prophylaxis. Conversely, in low-resource settings and countries, where infant screening is not standard care, individuals may die young even before diagnosis is confirmed, most frequently due to severe anaemia, which increases susceptibility to infections, such as bacterial sepsis and malaria (Inusa et al, 2019). This demonstrates the importance of early screening processes such as Hb electrophoresis. A result showing HbS and no normal HbA is required for definitive diagnosis (Lobitz et al, 2018).
Treatment of sickle-cell disease involves prescribing hydroxycarbamide and/or l-glutamine daily to reduce the rate of acute complications such as vaso-occlusive crises and managing these when they occur with medications, including pain relief. Blood transfusions can also be used to increase HbA, which subsequently improves oxygenation in severe anaemia, as well as reducing the proportion of HbS; these may be given as a simple top-up blood transfusion or as an exchange transfusion. Recent developments in bone-marrow transplant and gene editing trials have resulted in curing some people of sickle-cell disease (Inusa et al, 2019; Frangoul et al, 2021).
Case study
Presentation
Archie, who is 11 months old, is brought into a minor injuries unit by his mother after he had a fall outside their flat, attempting to run after his older brother. Archie had cut his knee on the concrete and had worried his mother due to the large amount of blood that had formed around him. On inspection, she did not find any open areas, other than the knee, and was struggling to stop the bleeding herself with a cold compress, but the wound continued to bleed even after several minutes.
History
This is the third time Archie attended the minor injuries unit – he had two similar incidents in the preceding few months since beginning to mobilise independently. Archie has also been having nosebleeds. When asked if there was a family history of bleeding disorders, his mother reported that her uncle had died from a bleed on the brain. Archie is not currently taking any medications and has a healthy appetite.
Physical examination
During examination of the wound it began to bleed again. Several older bruises on the arms and legs were also noted, along with some joint stiffness.
Initial interpretation
Based on family history and Archie's previous episodes of bleeding and bruising, several bleeding disorders could be considered. Further testing is required to identify where in the coagulation pathway Archie's blood abnormality can be placed.
In infants presenting with bruising, it is important to consider the location of bruises. If the bruising is not on the front of the body or overlying bone, or it is noted on babies who are not yet mobilising, this may flag up a safeguarding concern (Ward et al, 2013).
Further investigations
Archie has several tailored blood tests undertaken to determine the cause by excluding several possible underlying haematological conditions (differentials) resulting in excessive bleeding, as shown in Table 5.
Table 5. Archie's test results compared with results in haemophilia A, von Willebrand's disease, and vitamin K deficiency
| Test | Normal* result | Result in haemophilia A | Result in von Willebrand's disease | Result in vitamin K deficiency | Archie's result |
|---|---|---|---|---|---|
| Platelet count | 150–400 x109/L | Normal | Normal | Normal | 250 x 109/L |
| Bleeding time | 1–9 min | Normal | ↑ | Normal | 7 min |
| Prothrombin time (PT) | 11.5–13.5 s | Normal | Normal | ↑ | 12 s |
| Activated partial thromboplastin time (APTT) | 26–37 s | ↑+ | ↑+ | ↑ | 115 s |
| von Willebrand factor (VWF) | 50–200 IU/dL | Normal | ↓ | Normal | 100 IU/dL |
| Factor VIII concentrate (VIII:C) | 50–200% | ↓++ | ↓ | Normal | <1% |
Interpretation of findings
Archie has a family history of bleeding disorders, and his blood results show that his platelets are within the normal range. His bleeding time (assessing platelet function) is on the long side, but is still within normal range. Archie's PT is also within normal range, demonstrating that his extrinsic coagulation pathway is functioning appropriately. However, his APTT is elevated, indicating a defect in the intrinsic coagulation pathway involving factor VIII, IX, XI, and XII; he is therefore, unable to produce normal fibrin clots to stop bleeding. If both Archie's PT and APTT were raised, it would indicate an abnormality in the common pathway involving factors I, II, V, and X, such as vitamin K deficiency.
Further testing revealed that it was factor VIII that is abnormally low in Archie's blood. Based on Archie's signs and symptoms, family history and test results, he was diagnosed with severe haemophilia A, an X-linked recessive bleeding disorder predominantly affecting males (Khair, 2021).
Disease management
Replacement of the missing factor VIII via frequent intravenous infusions is key in the treatment of haemophilia A, with dose and frequency varying between individuals and over time (Isfordink et al, 2021). Factor VIII concentrates comprise multiple donors' plasma and therefore carry a higher risk of infection by blood-borne viruses, such as hepatitis and human immunodeficiency viruses (HIV), than products produced from single donations, such as red cells. This resulted in an epidemic of HIV and viral hepatitis in people with haemophilia. Fortunately, the introduction of viral inactivation of plasma-derived concentrates and increasing use of recombinant factor VIII replacement (a genetically engineered factor VIII, free of contaminating viruses, with an extended half-life) has almost eliminated new hepatitis viral infections in people with haemophilia (Isfordink et al, 2021). Trials with gene therapy are also in progress using adenovirus vectors containing the genes for factor VIII (Khair, 2021).
Treatment of acute bleeding is also key in haemophilia A management. World Federation of Haemophilia guidelines (Srivastava, 2020) recommend immediate initiation of treatment through the emergency department involving specialist health professionals.
Long-term complications of haemophilia A include chronic pain and repeated bleeding into the joints (haemarthropathy), which can lead to joint deformities and arthritis. Therefore, Archie should consider sports such as cycling, swimming and walking to strengthen his joints and muscles, and avoid contact sports such as rugby and martial arts to reduce the risk of internal bleeding (Srivastava, 2020).
Furthermore, the importance of familial support and involvement should not be overlooked as the diagnosis of chronic illness in infancy is often unexpected and can result in emotional turmoil. Health professionals should therefore provide tailored communication to families, educating them about haemophilia A and how to manage the condition. This includes developing individualised care plans, which can be given to teachers and other caregivers, explaining the importance of good oral hygiene, aid recognition of early signs and symptoms of haemorrhage, and what to do in these situations, such as replacement therapy at home if appropriate, or emergency hospital admission (Khair, 2021). Health professionals may also signpost the family to support groups of other effected individuals and their families, as well as provide further psychological support to enable the family to feel in control of the disease and its treatment, which subsequently improves quality of life (García-Dasí et al, 2016).
KEY POINTS
- The key components of blood are red blood cells, white blood cells, platelets and plasma
- Each component of blood is adapted to maximise its specific function
- Blood travels in vessels to supply the body's tissues with everything they need to facilitate life and to remove waste products
- If one or more components of the blood are affected, such that they cannot normally perform their role, this leads to blood disorders and disease
- Sickle-cell disease is an inherited recessive red-cell disorder, in which sickled haemoglobin is produced, which has a reduced life span and can block blood vessels
- Haemophillia A is an X-linked recessive bleeding disorder, which reduces factor VIII levels in the blood, so people with the disease will bleed for longer
CPD reflective questions
- What factors determine a person's blood group and why is it important to ensure that patients do not receive a transfusion of the wrong blood type?
- What has influenced the survival of the mutated gene resulting in sickle-cell disease and why is early diagnosis of sickle-cell anaemia important?
- How can health professionals effectively support families of infants diagnosed with haemophilia A, and why is this important?